Q-Chem es un potente software de química cuántica utilizado para cálculos ab initio de la estructura electrónica de sistemas moleculares. Diseñado para investigadores y científicos, Q-Chem ofrece una amplia gama de métodos teóricos, como Hartree-Fock (HF), Teoría del Funcional de la Densidad (DFT), métodos de perturbaciones (MP2) y Clúster Acoplado (CC), lo que lo hace ideal para estudios de estructuras moleculares, reactividad química y espectroscopía.
El software está optimizado para computación de alto rendimiento y cuenta con características avanzadas como cálculos de estados excitados mediante TDDFT y EOM-CC, simulaciones de solventes con modelos de solvatación continua, y acoplamiento cuántico-mecánico/mecánico-molecular (QM/MM) para el estudio de sistemas biológicos y materiales.

La versión 6.2 de Q-Chem, lanzada el 6 de mayo de 2024, introduce una variedad de características y mejoras significativas en el software de química cuántica. A continuación, se detallan algunas de las novedades más destacadas:

Q-Chem admite funciones LDA, GGA y meta-GGA, así como versiones híbridas, híbridas separadas por rangos y doblemente híbridas de GGA y meta-GGA. Las energías de un solo punto, las optimizaciones geométricas, los cálculos de frecuencia vibratoria y muchas otras propiedades se pueden evaluar para los estados fundamentales y para los estados excitados a través de DFT dependiente del tiempo.

Q-Chem ofrece herramientas de última generación para tratar los efectos de correlación de electrones, como la teoría de perturbaciones de Møller-Plesset y la teoría de grupos acoplados. Para los sistemas con una fuerte correlación, Q-Chem ofrece tratamientos especiales que incluyen CASSCF, teoría del enlace de valencia de grupos acoplados, CI seleccionado, RAS-CI, spin-flip y métodos variacionales 2-RDM.

Q-Chem proporciona un conjunto diverso de métodos para estudiar estados excitados electrónicamente: CIS, TD-DFT, NOCI, EOM-CC y ADC. Los sabores especiales de estos métodos cubren muchos tipos de estructuras electrónicas, lo que hace posible simular características espectroscópicas, transferencia de carga y energía y dinámicas no adiabáticas. Además, nuestro módulo de análisis de función de onda se puede utilizar para proporcionar más información sobre los estados excitados.
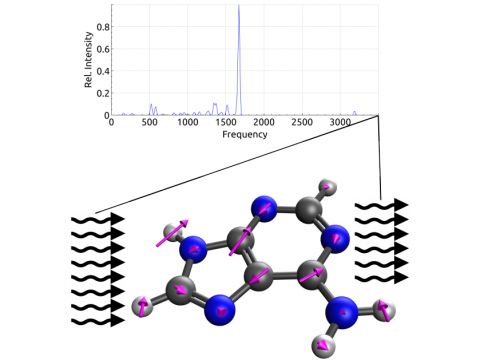

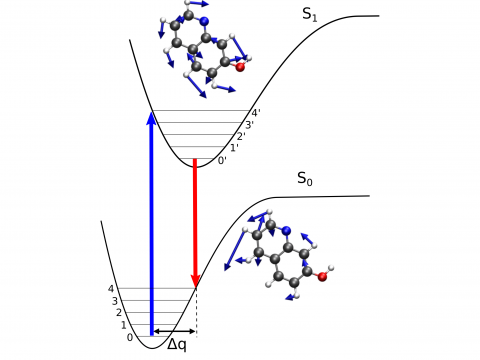
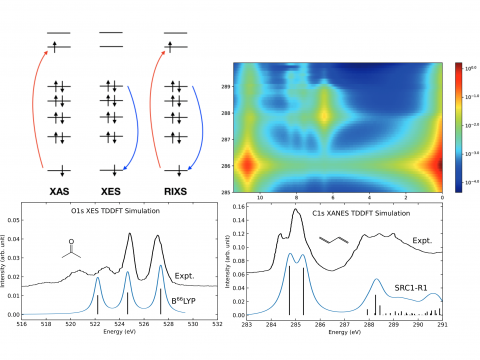


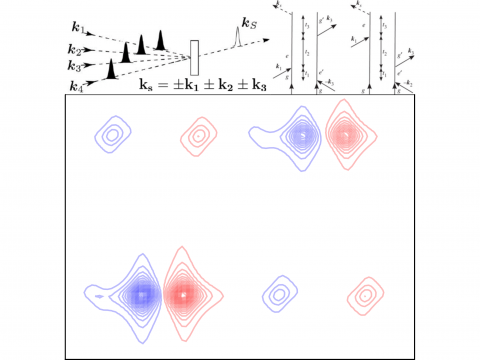
Q-Chem ofrece una variedad de herramientas para modelar diferentes tipos de espectros. Nuestras capacidades incluyen espectroscopía IR y Raman, espectroscopía UV-vis, espectroscopía de rayos X, espectroscopía de fotoelectrones, espectroscopía NMR y espectroscopía no lineal (como la absorción de dos fotones). Las características espectroscópicas se pueden estudiar utilizando muchos niveles diferentes de teoría, que van desde los métodos TDDFT hasta EOM-CC y ADC.


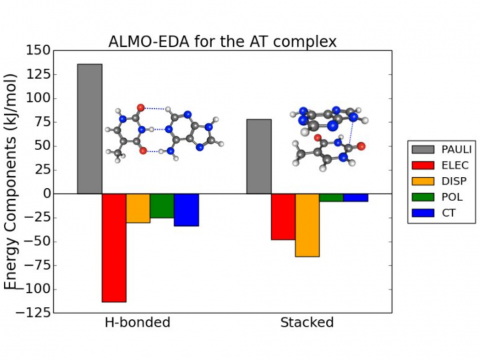
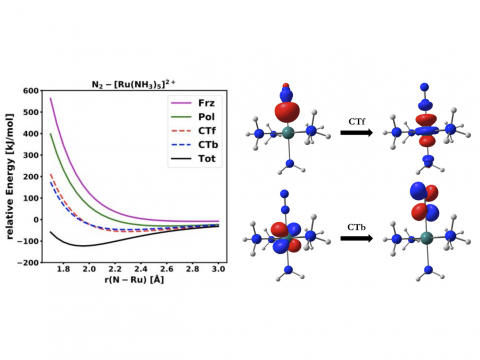
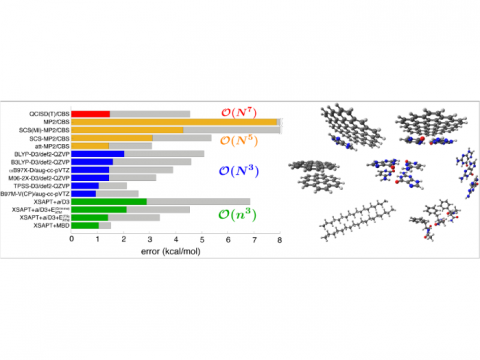
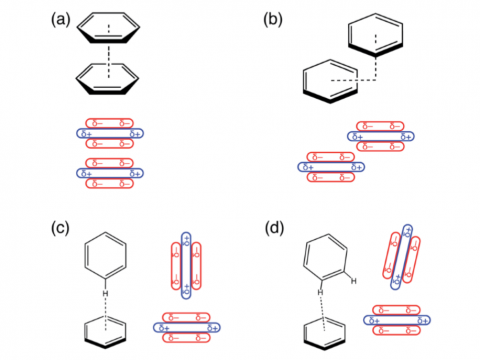
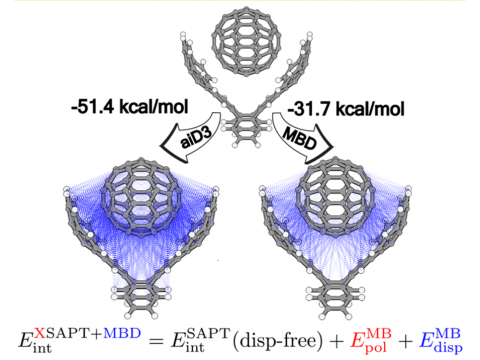
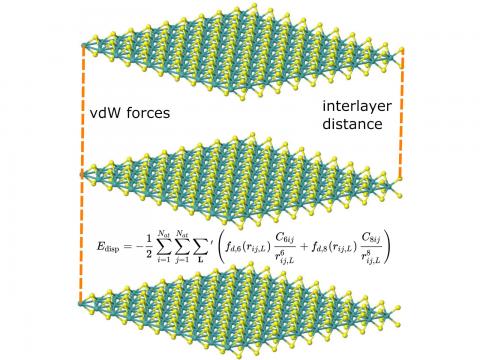
El análisis de descomposición de energía basado en orbitales moleculares absolutamente localizados proporciona un desglose de la energía de interacción total en términos físicos significativos, proporcionando información sobre la naturaleza de las interacciones intermoleculares y enlazadas. La teoría de la perturbación adaptada a la simetría (SAPT) y una versión extendida de muchos cuerpos de la misma (XSAPT) también están disponibles para calcular y analizar las interacciones intermoleculares.

Q-Chem proporciona métodos para la optimización de geometría, escaneos de superficie de energía potencial, búsquedas de estado de transición y seguimiento de coordenadas de reacción intrínseca, lo que lo hace ideal para estudios de reactividad química, termoquímica y cinética química.
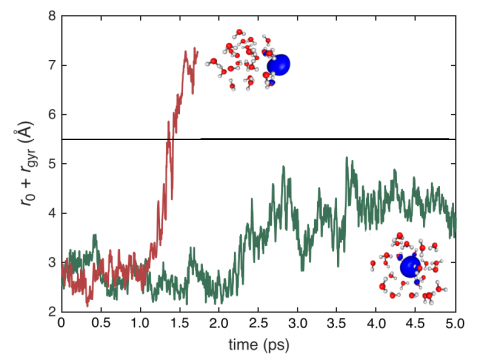

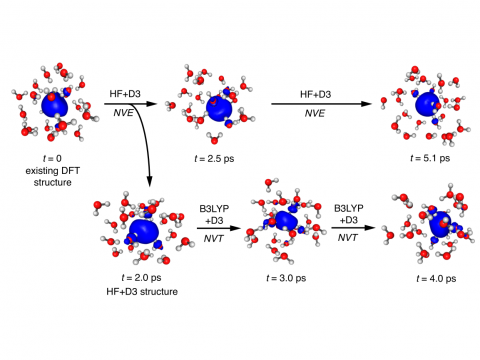
Q-Chem puede realizar dinámicas moleculares ab initio (AIMD), incluidos muestreos térmicos NVE y NVT, así como dinámicas moleculares casi clásicas (QMD). Estos enfoques se pueden utilizar para producir espectros de vibración e integrales de trayectoria ab initio. También incluimos una implementación del enfoque de salto de superficie con menor cantidad de interruptores (FSSH) de Tully para manejar de manera efectiva los sistemas no adiabáticos.
Haz clic aquí para conocer más del desarrollador de Q-chem
Q-Chem 6.2 | Fast, Accurate, Robust Chemistry Simulations | Q-Chem
Le recordamos que MultiON es distribuidor autorizado de Q-chem
Para más información sobre este u otros softwares especializados, entre en contacto con:
Luis Franco
Ejecutivo de Software Diverso
lfranco@multion.com
+52 (55) 55594050 Ext.118
¡Solicite su cotización ahora! Estaremos felices de orientarlo en su compra.
Haz clic aquí para conocer nuestros webinars gratuitos que impartimos mensualmente.
Descubra las últimas noticias sobre los softwares estadísticos y matemáticos que distribuimos aquí.
Q-Chem: facilita avances científicos a nivel mundial
Áreas de aplicación del Producto:
Física y Química